CIDP-Diagnose laut Leitlinien1,2
Spezifizierung der Diagnosekriterien nach EAN/PNS-Leitlinie
Bei CIDP-Varianten treffen nicht alle Kriterien der typischen CIDP zu, außerdem sind die Reflexe in den nicht betroffenen Gliedmaßen normal:
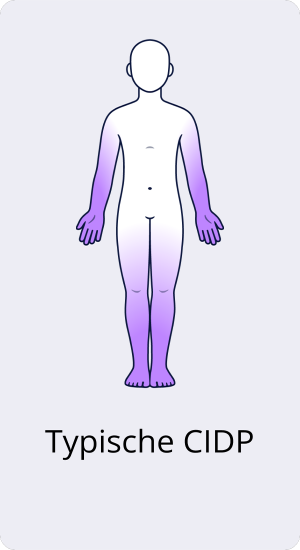
Typische CIDP
Die sogenannte typische CIDP liegt vor, wenn die Muskelschwäche beidseitig symmetrisch an Armen und Beinen auftritt, gekoppelt mit Gefühlsstörungen. Die Symptome schreiten entweder kontinuierlich oder schubförmig voran.
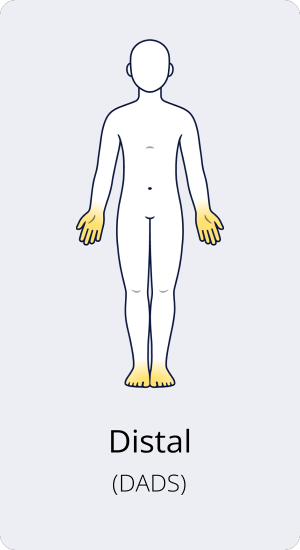
Distale CIDP oder DADS
Die distale CIDP oder DADS (distale, akquirierte demyelinisierende symmetrische Neuropathie) zeigt sich mit Sensibilitätsverlust in Händen und Füßen sowie einer Gangunsicherheit. Distal bezeichnet die vom Körperstamm weiter entfernten Teile der Gliedmaßen, also Hände und Füße. Tritt Muskelschwäche auf, ist sie in der Regel in den unteren Gliedmaßen (Füßen) ausgeprägter als in den oberen (Händen).
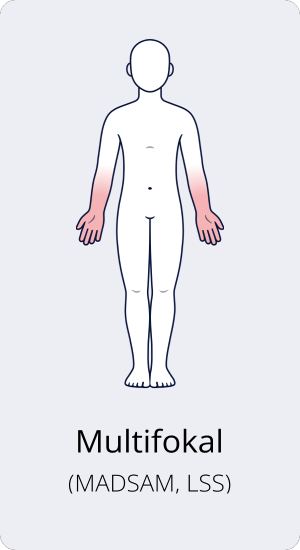
Multifokale CIDP oder MADSAM
Bei der multifokalen CIDP, auch Lewis-Sumner-Syndrom (LSS) oder MADSAM (engl.: multifocal acquired demyelinating sensory and motor neuropathy; dt.: multifokale erworbene demyelinisierende sensorische und motorische Neuropathie) genannt, treten die motorischen und sensorischen Symptome nicht symmetrisch auf beiden Körperseiten auf. In der Regel äußert sich die multifokale CIDP zuerst an den Armen, später können die Beine folgen. Häufiger als bei anderen Formen der CIDP sind auch Nerven im Kopfbereich betroffen, sodass z. B. Sehstörungen auftreten.
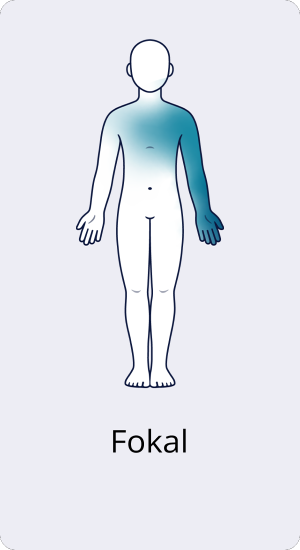
Fokale CIDP
Die fokale CIDP ist sehr selten und betrifft nicht ganze Gliedmaßen, sondern einzelne Nervengeflechte. Eines davon ist für die Motorik von Schulter- und Brustmuskulatur sowie die Motorik und Sensorik von Arm und Hand zuständig. Ein anderes, von der fokalen CIDP betroffenes Nervengeflecht versorgt die Gesäßmuskulatur, rückseitige Ober- und Unterschenkelmuskeln sowie sensorisch die Beinrückseite bis zum Fuß. Dementsprechend sind bei der fokalen CIDP Ausfälle in ebendiesen Regionen zu beobachten.
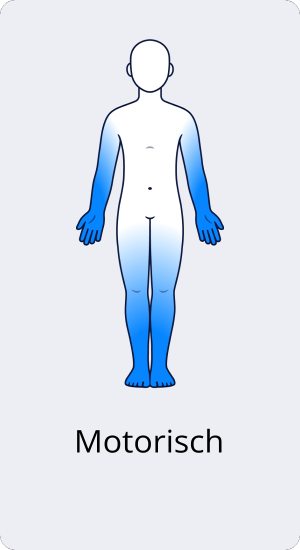
Motorische CIDP
Die rein motorische CIDP äußert sich durch eine beidseitige, symmetrische Schwäche der Extremitäten, also der Motorik. Die Gefühlswahrnehmung (Sensorik) ist nicht betroffen.
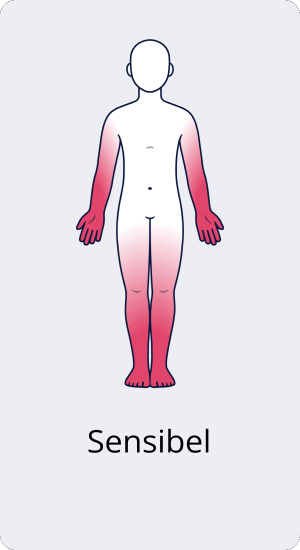
Sensible CIDP
Bei der rein sensiblen CIDP ist nur die Sensorik betroffen, es tritt keine Muskelschwäche auf. Durch die Beeinträchtigung der Sinneswahrnehmung treten allerdings neben Veränderungen des Hautgefühls auch Störungen des Gleichgewichts und Lagesinns und damit verbundene mögliche Unsicherheiten beim Gehen auf. Langfristige Untersuchungen haben gezeigt, dass die sensible CIDP häufig ein vorübergehendes klinisches Stadium darstellt, das bei etwa 70 % der Patient:innen in eine typische CIDP mit zusätzlicher Muskelschwäche übergeht.
Elektrophysiologische Parameter
Die EAN/PNS-Leitlinie gibt eine starke Empfehlung, die Elektrodiagnostik zur Bestätigung einer Verdachtsdiagnose CIDP basierend auf den klinischen Kriterien einzusetzen. Die Messungen können ein sehr gutes Bild über das Vorliegen und das Ausmaß der Nervenschädigungen vermitteln.
Elektrodiagnostische Kriterien für eine typische CIDP:
- In mindestens zwei Motornerven werden die Kriterien für Demyelinisierung erfüllt.
- Gefühlsstörungen in mindestens zwei Nerven.

die klinischen Kriterien und zudem
die elektro-diagnostischen Kriterien an einem Nerv erfüllt sind
oder der laut EAN/PNS empfohlenen Therapien Erfolg zeigt.



 ENG-Kriterien für Demyelinisierung
ENG-Kriterien für Demyelinisierung
In mindestens 2 Motornerven:
- Verlängerung der distal motorischen Latenz ≥ 50 % über ULN in zwei Nerven (Ausschluss Karpaltunnelsyndrom) oder
- motorische Nervenleitgeschwindigkeit herabgesetzt ≥ 30 % unter LLN in zwei Nerven oder
- Verlängerung der F-Wellen-Latenz ≥ 30 % ULN in zwei Nerven (≥ 50 %, falls die Amplitude des distalen negativen CMAP-Peaks < 80 % LLN) oder
- Fehlen der F-Wellen in zwei Nerven, falls die distalen negativen CMAP Peaks ≥ 20 % LLN+ ≥ 1 anderer demyelinisierender Parametera in ≥ 1 anderen Nerven oder
- partieller motorischer Leitungsblock: ≥ 50 % Reduktion der Amplitude des proximalen negativen CMAP-Peaks, verglichen mit Stimulation distal, falls der distale negative CMAP-Peak ≥ 20 % LLN in zwei Nerven oder in einem Nerven + ≥ 1 andere demyelinisierende Parameter in ≥ 1 anderen Nerven oder
- abnorme zeitliche Dispersion (> 30 % Zunahme zwischen dem proximalen und distalen negativen CMAP-Peak) in ≥ 2 Nerven oder
- Zunahme der Dauer des distalen CMAP (Intervall zwischen Beginn des ersten negativen Peaks und Rückkehr zur Grundlinie des letzten negativen Peaks) in ≥ 1 Nerv (N. medianus ≥ 6.6 ms, N. ulnaris ≥ 6.7 ms, N. peroneus ≥ 7.6 ms, N. tibialis ≥ 8.8 ms) + ≥ 1 andere demyelinisierende Parametera in ≥ 1 anderen Nerven
ENG: Elektroneurographie; CMAP: Muskelantwortpotenzial; ULN: oberer Grenzwert; LLN: unterer Grenzwert.
a. Jeder Nerv, der eines der Kriterien a–g erfüllt.
-

Liquoruntersuchung
Im Liquor lassen sich bei einem Großteil der CIDP-Patient:innen eine leicht erhöhte Zellzahl (in der Regel < 10) und eine deutlich erhöhte Eiweißkonzentration nachweisen. Dies sind aber keine sicheren Diagnosekriterien.
-

Blutwerte
Regulär erhobene Blutwerte sind bei CIDP in der Regel diagnostisch nicht hilfreich. Es lassen sich aber bestimmte Antikörper nachweisen, die zur Diagnosestellung eingesetzt werden können. Dieser Nachweis wird vorwiegend als Ausschlussdiagnose gegenüber anderen Polyneuropathien eingesetzt.
- Eine erhöhte Immunglobulin-Konzentration im Blut, auch monoklonale Gammopathie unklarer Signifikanz (MGUS), kann ein Hinweis auf eine maligne Erkrankung sein, der unbedingt abgeklärt werden muss.
- Der Nachweis von Antikörpern gegen Proteine der Ranvier’schen Schnürringe (nodale und paranodale Proteine) wie Anti-NF155, Anti-CNTN1 und Anti-Caspr1 kann eine Verdachtsdiagnose bestätigen, wenn andere Diagnoseparameter keine eindeutige Zuordnung erlauben.
Weitere diagnostische Optionen
-

Bildgebende Verfahren
Die Magnetresonanztomographie (MRT) mit T2-Wichtung kann eingesetzt werden, um Nervenläsionen, insbesondere an den Nervenwurzeln, nachzuweisen. Diese sind typisch für CIDP.
Die Sonographie (Ultraschall) kann eingesetzt werden, um die verschiedenen Neuropathien voneinander abzugrenzen. Somit können anatomische Veränderungen einzelner Nerven wie Verdickungen festgestellt werden, die bei einer Schädigung durch Ansammlung von Entzündungsflüssigkeit im Nervengewebe entstehen.
-

Neurobiopsien
In Biopsien kann zwar die Demyelinisierung der Nervenzellen direkt nachgewiesen werden, sie sind aber recht aufwendig und invasiv. Sie werden nur eingesetzt, wenn klinische und elektrodiagnostische Kriterien, bildgebende und Laboruntersuchungen keine eindeutige Diagnose zulassen. Neurobiopsien sollten nur in spezialisierten Zentren durchgeführt werden.
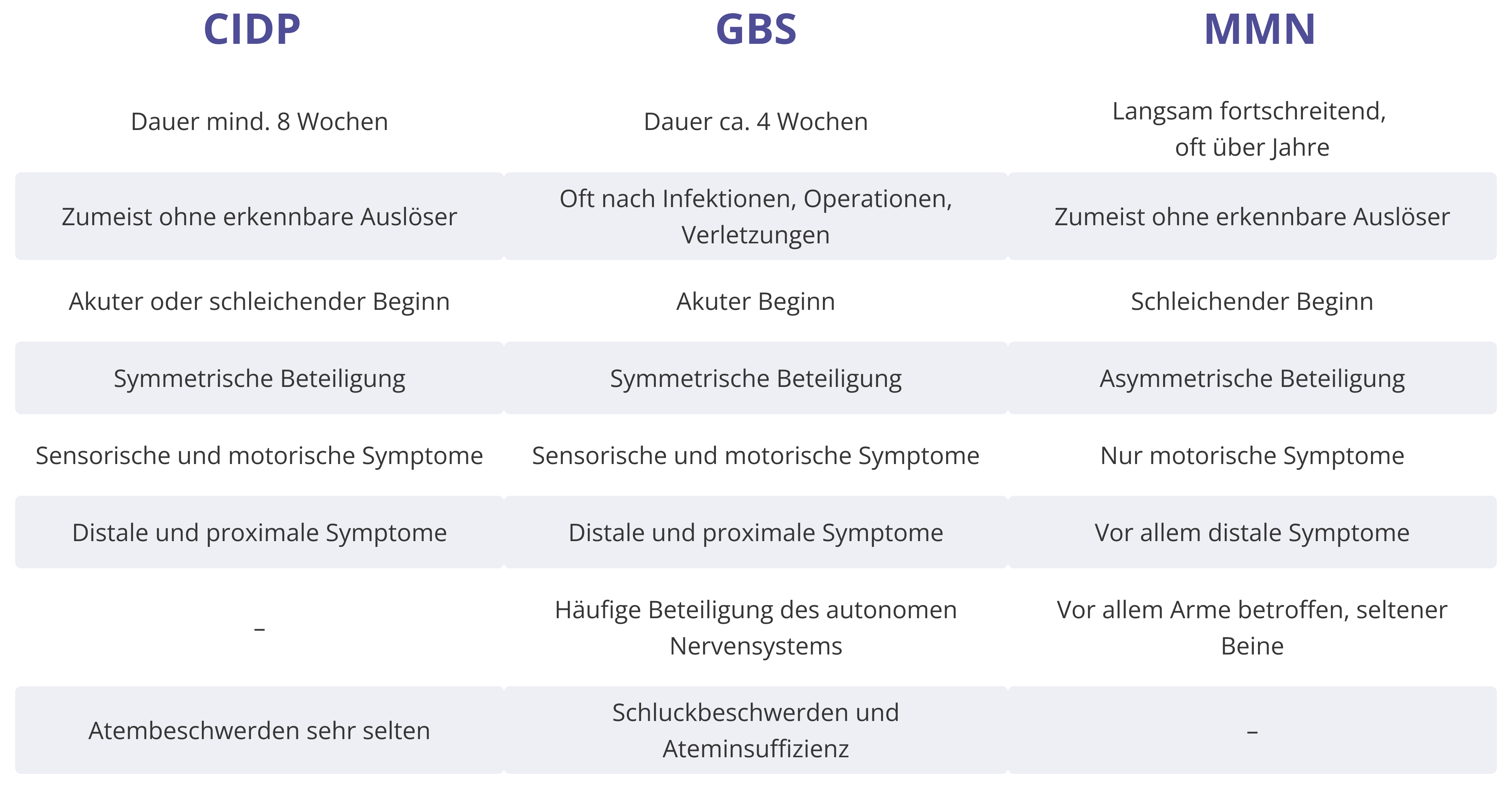
Abgrenzung entzündlicher Polyneuropathien CIDP, GBS und MMN
Quellen
- Van den Bergh PYK et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force-Second revision. Eur J Neurol. 2021;28(11):3556–3583.
- Grimm A, et al. DGNeurologie. 2022;5 (2):114–125.
- Markowitz JA. et al. Neurology. 2008;71:e74–e78.
C-APROM/DE/IG/0038